
14/12/2021 - Press release
This new resource, developed by the GPCR Drug Discovery Group at the Hospital del Mar Medical Research Institute, provides a three-dimensional analysis of the movements of COVID-19 proteins. This can help researchers understand how they work and develop new treatments and vaccines
The tool, available online to all researchers, offers a large number of simulations of how these proteins work, as well as resources for predicting how their function could change in relation to mutations that may occur in the structure of this coronavirus
The scientists behind the initiative used more than 360 gigabits of data to develop it. To date, it is the only database for studying SARS-CoV-2 that combines protein simulations with mutation data.
Researchers now have a new tool available for tackling SARS-CoV-2, analysing this virus, and finding new ways to fight COVID-19, namely the SCoV2-MD database (available at www.scov2-md.org). The resource houses detailed atomic, dynamic and 3D information on all known 3D-structured proteins in this coronavirus. In total, it contains 360 gigabits of data covering the majority of the 29 proteins: four structural, sixteen non-structural, and nine accessory proteins. Nucleic Acids Research has published a paper on how it works, which it considers to be one of the most prominent articles ever published in the journal.
Proteins are basic molecules in the functioning of cells. In the case of SARS-CoV-2, the coronavirus responsible for COVID-19, they are responsible for its ability to both infect humans and propagate. This is the case of the so-called spike protein, which makes up the characteristic crown that gives this type of virus its name. The new database, developed by the GPCR Drug Discovery Group at the Hospital del Mar Medical Research Institute (IMIM-Hospital del Mar), in collaboration with the Biophysics Institute (CNR-IBF) of the National Research Council of Italy, the Paul Scherrer Institute of Switzerland, and Dompé Farmaceutici of Italy, provides an unprecedented level of detail on the structure and functioning of the virus, as well as predicting its evolution through the different mutations that it has undergone and will undergo in the future.

From left to right: Jana Selent and Mariona Torrens-Fontanals.
A tool available to all researchers
This tool, one of the most powerful ever created that combines simulations of the 3D structure of proteins with data on mutations in the virus responsible for COVID-19, is available to any researcher, online and free of charge. By connecting, without the need to use a specific app, they can see the structure of SARS-CoV-2 proteins at the atomic level, thanks to the analysis carried out by the creators of the database from the information they generated themselves together with that available in different public repositories. For this purpose, computational tools for simulating molecular dynamics have been used to make predictions of how each atom of the protein will act according to the laws of physics, in order to build a three-dimensional model of the molecule's behaviour. The accumulated simulations, 252 in total, also address the impact of known coronavirus mutations on the proteins.
"What is really novel is that we use dynamic data combined with the evolution of the virus to predict its impact on protein function", explains Jana Selent, an IMIM-Hospital del Mar researcher and lead author of the paper on this new tool. To do this, "In addition to protein behaviour simulations, the information available on the genetics of the virus has been taken into account to calculate and predict the impact of mutations", adds Toni Giorgino, co-lead author of the article. This makes SCoV2-MD a useful tool for visualising how these SARS-CoV-2 mutations will affect its ability to transmit and infect human cells through the changes produced in their constituent proteins.
Meanwhile, this new tool can also help researchers develop new treatments and vaccines against COVID-19. "Watching the simulations allows us to see and understand how it behaves, how it works, and which parts of the protein's structure are important and possible targets for new treatments", says Mariona Torrens-Fontanals, also a researcher at IMIM-Hospital del Mar and first author of the study. It even makes it possible to predict whether coronavirus mutations could affect the ability of the antibodies generated by COVID-19 vaccines to recognise the virus and activate the immune system against it.
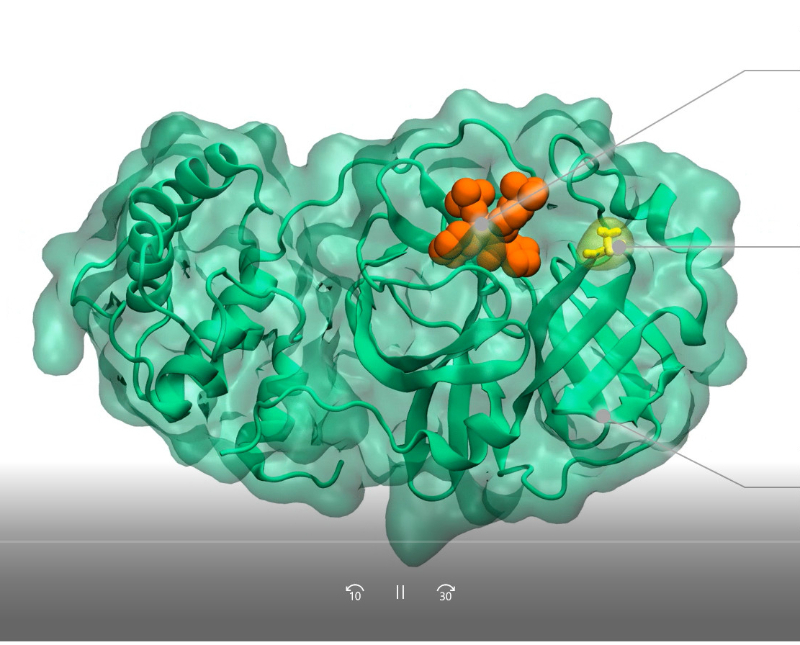
The video shows that by combining protein structural dynamics and virus evolution data, SCoV2-MD allows studying the impact of specific mutations (e.g. T24A in the SARS-CoV-2 main protease) on the binding of antiviral compounds (e.g. the inhibitor ML188).
Reference article
Mariona Torrens-Fontanals, Alejandro Peralta-García, Carmine Talarico, Ramon Guixà-González, Toni Giorgino, Jana Selent, SCoV2-MD: a database for the dynamics of the SARS-CoV-2 proteome and variant impact predictions, Nucleic Acids Research, 2021; https://doi.org/10.1093/nar/gkab977
© Institut Hospital del Mar
d'Investigacions MèdiquesLegal Notice and Privacy Policy | Cookie Policy | Site Index | Accessibility | Find Us | Contact
